




It is impossible to overstate the value of drug trials in advancing healthcare services. Patients’ quality of life and longevity can be increased by new medications and treatments. Although more clinical trials are required, the government is also working to protect the subjects’ rights and safety and raise the quality of the studies conducted in India to meet international standards. India has served as a hub for several multi-center studies. The rules for conducting clinical trials in India are set forth by the Central Drugs Standard Control Organization (CDSCO), which is led by the Drug Controller General of India (DCGI). This growth might be attributed to the way trials were conducted, Indian laws, and the quality of the data produced. It is imperative that going forward, all clinical trials carried out in India abide with Schedule Y of the Drugs and Cosmetics Act and the International Conference of Harmonization’s Good Clinical Practices Guidelines (ICH-GCP).
The regulations regarding the clinical trials in India are outlined under Schedule Y, along with the prerequisites and rules for the manufacturing and import of novel medications for retail sale or clinical trials. It covers the steps involved in making an application to conduct clinical trials, as well as the duties of the sponsor, investigators, and Independent Ethics Committee.
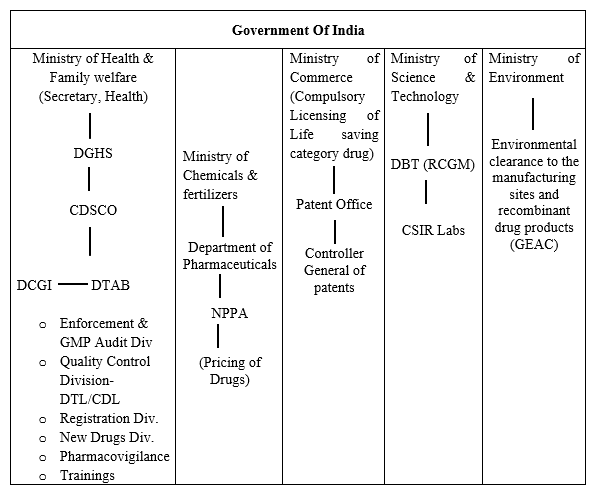
Drug Regulatory System of India
Clinical trials and drug regulations are both governed by the strict regulation in India. The diagram below provides an overview of the regulatory system:
What Do We Mean By Clinical Trials?
A clinical trial is a research study that examines whether a novel medical procedure or innovative application of an already-proven procedure will be a more effective means of disease prevention, detection, diagnosis, or treatment. Any novel medication must pass preclinical testing in order to begin a clinical trial. Preclinical research includes experiments on animal populations and in vitro (also known as test-tube or laboratory) research. To gather preliminary information on the research medication’s effectiveness, toxicity, and pharmacokinetics, a wide variety of doses of the study drug are administered to animal subjects or to an in-vitro substrate.
Anyone planning a clinical trial in India of a novel therapy must carefully assess the objectives, issues, risks, and advantages of each therapy, and the alternatives they select must be both morally and scientifically justifiable.
Why Does A Clinical Trial Need To Be Conducted?
The “principle of essentiality,” as defined by the Indian Council of Medical Research also known as ICMR, serves as the foundation for every clinical research. Simple words, a clinical study is carried out because it is necessary. Scientists and governments would be more than willing to adopt other approaches to assess new drugs if those methods were accessible. The clinical trial is still the most thorough technique to evaluate novel medications today, even though many initiatives are being investigated to lower the number of individuals exposed to new clinical trials.
Therefore, when a physician offers to enrol a patient in a clinical trial, he is asking for the patient’s participation in an investigation to further medical knowledge. The goal is to provide patients with access to better medications in the future.
India as a Hotspot for Clinical Trial
Clinical research is not a new concept for India. Two ancient texts, the Charaka Samhita (a medical textbook) and the Sushruta Samhita (a surgical textbook), written in 200 B.C. and 200 A.D., respectively, demonstrate India’s longstanding expertise in medical study.
As early as 200 B.C., the Charaka Samhita (a course reading on medicine) and the Sushruta Samhita (a coursebook on a medical technique) were written. Additionally, the years 200 A.D. individually demonstrate India’s longstanding expertise in clinical research.
Over the past few decades, clinical pharmaceutical preliminary exams have grown enormously in India. Outside of the US, India was the second-most popular country to oversee clinical preliminary exams in 2009. To strengthen the clinical preliminary in India, many associations have provided numerous unusual guidelines related to the safe clinical preliminary. The primary legal authority in India for approving clinical trials as well as for marketing and assembling medications is the Drug Controller General of India (DCGI). The clinical research sector in India is estimated to be worth over INR 3500 million and is growing by 10-12% yearly. India is the top country with a hereditarily respectable population mix and the one where medical school prerequisites are the easiest to complete.
The Indian government has made several major strides to ensure the safety of trial participants, and it is presently mandatory to register all clinical studies with the Clinical Trial Registry of India (CTRI).
In order to safeguard the rights of trial subjects, the Supreme Court of India (SC) suggested stricter regulations for the conduct of clinical trials in September 2013. The Swasthya Adhikar Manch (Health Rights Forum), a non-governmental organisation that had documented irregularities and unethical practises in clinical trials conducted in the state of Madhya Pradesh, filed a public interest litigation (PIL) against the Union of India in the case of Swasthya Adhikar Manch v. Union of India [(Civil) No. 33 of 2012], the issues in this particular case includes, signing up volunteers without getting sufficient informed permission and not paying them enough in case they got hurt or died during the experiment. A review of the roughly 400 trials that were conducted in the five years between 2007 and 2012 was also requested in the PIL. The Central Drugs Standard Control Organization (CDSCO), India’s regulatory body, implemented a number of measures in response to the SC’s observations and directives, some of which have been blamed for a significant decrease in the number of clinical trials being conducted in India, with both domestic and foreign drug companies moving to alternative clinical trial sites.
Report of Parliamentary Standing Committee on Health
The Parliamentary Standing Committee on Health made a statement regarding test laboratories on September 12 of 2022. According to the committee, India has only 18 certified medical device testing labs that have been approved by CDSCO[1], which is egregiously insufficient given the size of the nation.
Prof. Ram Gopal Yadav, M.P., delivered the 138th Report to the Rajya Sabha on the topic of “Medical Devices: Regulations and Control.” Only 18 certified Medical Device Testing Laboratories have been licenced by CDSCO, according to the report from the committee, which is glaringly insufficient given the population of the nation. The Committee is the opinion that having sufficient common infrastructure, including accredited laboratories in various regions of the nation for standard testing, would significantly encourage local manufacturers to have their products tested for standards. Such measures would also help in reducing the cost of production, which in turn will improve the availability & affordability of medical devices in the domestic market.
Additionally, a strong IT-enabled feedback-driven post-market surveillance system and medical device registry were advised, especially for implants, to provide patient traceability and the capacity to evaluate the effectiveness of the implant.
According to the report, the Committee has advised the Ministry to collaborate with State governments and provide the local medical device officers with the necessary training. The Committee has also advised the Ministry to devise a system for regularly designating State Medical personnel as Medical Device or Medical Device Testing Officers in order to effectively carry out the legislation’s mandate. The Committee thinks that because the industry is expanding so quickly, the government should not continue to allow pharma experts to regulate medical devices. Instead, it is time for qualified and well-trained medical device officers to enforce medical device regulations at the local level in order to support the nation’s medical device industry.
The Ministry should let the new regulator to collaborate with organisations like IISC, CSIR, DRDO, and a network of IITs to evaluate medical devices for efficacy and safety, according to the report’s recommendation. The Committee was certain that these institutions should be employed to examine medical devices for their electrical, electromagnetic, and biological components since they had high-tech labs. The Committee also suggests that more money be invested in raising the requirements for these labs’ standards.
Current Regulatory Authorities for Clinical Trials
The International Conference on Harmonization (ICH), which is governed by the United States, Europe, and Japan, now controls how clinical trials are conducted. Clinical research is covered by a variety of regulations in India. Despite the fact that there are several laws in place, the Indian Council of Medical Research (ICMR) Act of 1947 is crucial for clinical trials.
The following is a list of the laws that regulate clinical trials in India:
- Drugs and Cosmetics Act of 1940 (Schedule Y)
To guarantee that pharmaceuticals and cosmetics marketed in the nation are secure, efficient, and in compliance with fundamental quality requirements, this act, which was initially created in 1940, controls the import, production, and distribution of drugs in the nation. To guarantee higher safety, effectiveness, and medication quality, it is regularly revised and contains Chapters, Rules, and Schedules. The main regulation governing clinical research in the nation is Schedule Y, together with Rules 122A, 122B, 122D, 122DA, 122DAC, and 122E. All clinical research that is covered under Schedule Y is required by law to adhere to the necessary guidelines. There are 12 appendices, including templates for informed consent forms, clinical trial protocol types, templates for ethics committee (EC) approval, and a format for significant adverse event (SAE) reporting.
- Ethical Guidelines of the Indian Council of Medical Research of 2006
The updated ICMR standards, known as the “Ethical Guidelines for Biomedical Research on Human Participants,” were published in 2006 and are still in effect as of this writing. A further revision is anticipated in 2017. This recommendation addresses two major facets of clinical research: general guidelines that must be followed and recommendations for unique fields of study (e.g., research in children or herbal research). Both of these documents must be understood by researchers in order for them to follow both of theme’s instructions and needs.
- Indian Good Clinical Practice Guideline of 2001
In 2001, the CDSCO published a good clinical practise (GCP) guideline that aimed to be India-specific, however unlike the ICH GCP guideline, it has not been updated since.
- Indian GCP Guidelines
One of the oldest and most persistent traditions in the history of medicine is where the Good Clinical Practice (GCP) law gets its roots from. The design, conduct, termination, audit, analysis, reporting, and recording of biological research involving human participants are all covered by a set of rules known as “good clinical practise.”
The core principle of GCP is that, while doing research on humans, the needs of science and society should never come before those of the study subject. It tries to make sure that the investigations are morally and scientifically sound, and that the clinical characteristics of the pharmaceuticals under study are accurately described. The two guiding concepts of the rules are the preservation of human subjects’ rights and the veracity of biomedical data collected. These recommendations were developed considering the Ethical Standards for Biomedical Research on Human Subjects published by the Indian Council of Medical Research (ICMR) as well as WHO, ICH, USFDA, and European GCP guidelines.
- New Drugs And Clinical Trials Rules Of 2019
In accordance with the authority granted by subsection (1) of section 12 and subsection (1) of section 33 of the Drugs and Cosmetics Act, 1940, the New Drugs & Clinical Trials Rules, 2018 were published in the Indian Gazette. These regulations will be applicable to NDs, INDs for human use, CT, BA, BE, and ethics committee regulation pertaining to CT, BA/BE study and biomedical health research.
The definition of new pharmaceuticals has been updated to include xenografts, stem cells, gene therapy products, innovative drug delivery systems (NDDS), living modified organisms, monoclonal antibodies, and more.
The application for a CT of a new medicine that has already been authorised and commercialised in a nation must be processed within 90 working days.
The 13 chapters (which contain 107 regulations) and 8 schedules that make up the new rules form its framework. All novel pharmaceuticals, novel investigational medications for human use, clinical trials, bioequivalence & bioavailability studies, and ethics committees will be subject to the new regulations. Part XA and Schedule Y of the 1945 Drugs and Cosmetics Rules will be replaced by the new regulations, which take effect right away. Schedule Y, prior rules, and medications for veterinary use will still remain in effect.
Academic clinical trials are defined by the New Rules, 2019 as “clinical trials of a drug already approved for a certain claim initiated by any investigator, academic/research institution for a new indication, new route of administration, new dose/new dosage form, where the results of such a trial are intended to be used only for academic/research purposes & not for seeking approval of the Central Licensing Authority/regulatory authority of any country for m
The following are some key considerations for academic clinical trials:
- Only for authorised medications
- CT performed by a researcher, academic institution, or research centre
- Can be done for a novel indication, a novel route, a novel dose, or a novel dosage form.
- Results should only be used for academic or research purposes; not for profit. In no nation may data be used to request approval.
- The Central Licensing Authority (CLA) must answer to EC’s request for clarification within 30 days (or deemed that no approval is needed)
- In accordance with the ICMR Guidelines for Biomedical Research on Human Participants, medical management and compensation are appropriate.
- Academic CTs must follow the EC-approved CT procedure and the ethical guidelines outlined in the ICMR Guidelines for Biomedical Research on Human Participants.
The Ministry of Health & Family Welfare has changed the guidelines. In a notice dated August 31, 2021, the Ministry published the New Drugs and Clinical Trials (Amendment) Rules, 2021. The definitions of bioavailability and a bioequivalence research centre are altered by the Rules, which update the New Drugs & Clinical Trials Rules, 2019, respectively. The following is the updated definition under Rule 2(1)(g):
Bioavailability & bioequivalence study centre means a centre created/established to undertake bioavailability study or bioequivalence study of a drug for either clinical part or analytical part or for both clinical and analytical part of such study.
The following rules under the New Drugs and Clinical Trials Rules of 2019 should be understood:
- Rule 2(1)(a) talks about – academic clinical trial
- Rule 2(1)(k) talks about – clinical trial protocol
- Rule 2(1)(l) talks about – clinical trial site
- Rule 2(1)(o) talks about – Ethics Committee
- Rule 2(1)(p) talks about – Good Clinical Practices Guidelines
- Rule 2(1)(q) talks about – global clinical tria
- Rule 3 talks about – Central Licencing Authority
- Rule 6 talks about – Requirement of the Ethics Committee
- Rule 7 talks about – Constitution of Ethics Committee for clinical trial
- Rule 8 talks about – Registration of Ethics Committee relating to clinical trial, bioavailability & bioequivalence study.―
- Rule 9 talks about – Validity period of registration of Ethics Committee for clinical trial
- Rule 10 talks about – Renewal of registration of Ethics Committee for clinical trial
- Rule 12 talks about – Proceedings of Ethics Committee for clinical trial
- Rule 13 talks about – Maintenance of records by Ethics Committee for clinical trial
- Rule 14 talks about – Suspension/cancellation of registration of Ethics Committee for clinical trial
- Rule 19 talks about – Clinical trial of new drug or investigational new drug
- Rule 20 talks about – Oversight of clinical trial site
- Rule 21 talks about – Application for permission to conduct clinical trial of a new drug/investigational new drug
- Rule 22 talks about – Grant of permission to conduct clinical trial
- Rule 23 talks about – Permission to conduct clinical trial of a new drug/investigational new drug as part of discovery, research & manufacture in India
- Rule 24 talks about – Permission to conduct clinical trial of a new drug already approved outside India
- Rule 25 talks about – Conditions of permission for conduct of clinical trial
- Rule 26 talks about – Validity period of permission to initiate a clinical trial.
- Rule 27 talks about – Post-trial access of investigational new drug or new drug
- Rule 28 talks about – Academic clinical trial
- Rule 29 talks about – Inspection of premises relating to clinical trial
- Rule 30 talks about – Suspension or cancellation of permission to conduct clinical trial.
- Rule 39 talks about – Compensation in case of injury or death in clinical trial or bioavailability/bioequivalence study of new drug or investigational new drug.
- Rule 40 talks about – Medical Management in clinical trial or bioavailability & bioequivalence study of new drug/investigational new drug.
- Rule 41 talks about – Consideration of injury/death/permanent disability to be related to clinical trial/bioavailability and bioequivalence study.―
- Rule 42 talks about – Procedure for compensation in case of injury/death during clinical trial, bioavailability & bioequivalence study.
- Rule 43 talks about – Medical management & compensation for injury or death relating to biomedical & health research overseen by an Ethics Committee for biomedical & health research as referred to in Chapter IV
- Rule 52 talks about – Application for permission to manufacture of new drug/investigational new drug for clinical trial or bioavailability and bioequivalence study/for examination, test and analysis
- Rule 53 talks about – Grant of permission to manufacture new drugs or investigational new drugs for clinical trial or bioavailability/bioequivalence study, or for examination, test and analysis.
- Rule 54 talks about – Validity period of permission to manufacture of new drug/investigational new drugs for clinical trial or bioavailability & bioequivalence study, or for examination, test and analysis.
- Rule 56 talks about – Licence to manufacture new drugs/investigational new drugs for clinical trial or bioavailability/bioequivalence study/for examination, test & analysis under the Drugs and Cosmetics Rules, 1945
- Rule 57 talks about – Inspection of new drugs/investigational new drugs manufactured for clinical trial/bioavailability & bioequivalence study/for examination, test and analysis.
- Rule 59 talks about – Application for permission to manufacture unapproved active pharmaceutical ingredient for development of pharmaceutical formulation for test/analysis or clinical trial or bioavailability and bioequivalence study.
- Rule 60 talks about – Grant of permission to manufacture an unapproved active pharmaceutical ingredient for development of pharmaceutical formulation for test/analysis or clinical trial or bioavailability and bioequivalence study
- Rule 61. Validity period of the permission to manufacture an unapproved active pharmaceutical ingredient & its formulation for test/analysis/clinical trial or bioavailability and bioequivalence study.―
- Rule 62 talks about – Suspension/cancellation of permission to manufacture an unapproved active pharmaceutical ingredient for development of formulation for test/analysis or clinical trial or bioavailability and bioequivalence study.
- Rule 64 talks about – Licence to manufacture an unapproved active pharmaceutical ingredient for the development of formulation for test/analysis/clinical trial/bioavailability & bioequivalence study under the Drugs & Cosmetics Rules, 1945
- Rule 65 talks about – Inspection of manufacturer of an unapproved active pharmaceutical ingredient for development of formulation for test or analysis/clinical trial or bioavailability and bioequivalence study.
- Rule 67 talks about – Application for import of new drug/investigational new drug for clinical trial/bioavailability or bioequivalence study/for examination, test and analysis.
- Rule 68 talks about – Grant of licence for import of new drug/investigational new drug for clinical trial/bioavailability/bioequivalence study or for examination, test and analysis.
- Rule 69 talks about – Validity period of licence for import of new drugs for clinical trial/bioavailability/bioequivalence study or for examination, test and analysis.
- Rule 91 talks about – Application for permission to manufacture an unapproved new drug but under clinical trial, for treatment of patient of life threatening disease.―
- Rule 92 talks about – Grant of permission to manufacture an unapproved new drug but under clinical trial, for treatment of patient of life threatening disease.
- Rule 94 talks about – Inspection of unapproved new drug but under clinical trial manufactured for patient of life threatening disease.
Stages of Regulatory Approvals in the Process of Drug Development
The following is the stage wise regulatory approval for clinical trials in India:
Stage 1: Drug Discovery
Stage 2: Pre-Clinical Testing
Stage 3: Permission to Conduct Clinical Trial
Stage 4: Launch of New Drug
Stage 5: Registration of New Drug
Stage 6: Human Clinical Trial
Stage7: Post Marketing Surveillance
What Are The Legal Issues Involved In Making A Human Subject To Clinical Trials In India?
Schedule Y of the Drugs & Cosmetics Rules states:
Without the Drugs Controller General of India’s approval, new chemical entities cannot be given to volunteers in clinical research (DCGI). You can get this approval by submitting a clinical study application to the DCGI.
The application must include:
- A plan for the investigation,
- The Informed Consent Document’s draught,
- A list of potential researchers who have consented to take part in the study, and according to Schedule Y of the Drugs & Cosmetics Rules, background information regarding the substance is required.
For the majority of investigatory medications, getting approval for a clinical study takes close to 12 weeks. For medications that are particularly important to the nation’s healthcare needs or those that can be viewed as contentious, the time frame may be extended since these are likely to be sent to the Indian Council of Medical Research for opinions. A “Test-Import License” must also be requested in order to import clinical materials. Regulations 33 and 34 as well as the clauses in Part X-A of the rules control the import and manufacturing of clinical trial supplies.
The process for requesting marketing authorisation is determined by the new drug’s status, which generally falls into one of three categories:
- If phase III or confirmatory trials are carried out to gather data about the efficacy and safety of the drug in a sufficient number of patients, in comparison with a standard drug or a placebo, in order to confirm efficacy and safety claims made in the product monograph, that is sufficient for newly discovered drug substances that are already approved/marketed in other countries.
- New pharmacological compounds that have been identified but have not been licenced or commercialised in other nations are given “phase lag” approval for clinical studies. This implies that a phase I trial can only be approved if the medicine has already passed phase I and entered phase II in other nations. Similar to other nations, phase II in India is only permitted if phase II there has ended and phase III has started.
What Are The Consequences If A Human Subject Dies Under Clinical Trials?
Compensation in case of damage or death during a clinical experiment, under 122-DAB –
- If the subject sustains a harm while participating in the clinical study, free medical care will be provided for as long as necessary or until it is determined that the injury is unrelated to the experiment, whichever comes first.
- If a trial subject sustains an injury that is connected to the clinical trial, the subject will also be entitled to financial compensation, as per the order of the licencing authority described in clause (b) of Rule 21, which will be in addition to any costs associated with the subject’s medical care. If there is no lasting damage, the amount of compensation must be proportionate to the subject’s non-permanent damage and salary loss.
- According to the Licensing Authority’s order, which is defined in clause (b) of Rule 21, in the event that a subject involved in a clinical trial passes away, his or her nominee(s) would be entitled to financial compensation, which would be in addition to any costs associated with the subject’s medical care.
- The sponsor of the clinical trial is responsible for covering the costs of medical care and monetary compensation in the event that a trial participant is injured or dies during the course of the study.
- The following conditions shall be deemed to constitute clinical trial-related injuries or deaths, and the subject or his/her nominee(s), as applicable, shall be entitled to financial compensation for such injuries or deaths:
- Adverse impacts of the product(s) under examination.
- Violation of the approved protocol, carelessness on the part of the sponsor, his agent, or the investigator, as well as a violation of Drugs and Cosmetics Rules of 1945.
- Failure of the experimental product to have the desired therapeutic impact when, despite the availability of conventional treatment, the patient did not get it in accordance with the clinical study protocol.
- Using a placebo in an experiment where the individual was eligible for conventional care but it wasn’t given to them in accordance with the protocol for the clinical trial;
- Adverse drug interactions that prevent the need for routine care yet are necessary for following a procedure that has been approved;
- If a parent’s involvement in a clinical trial causes harm to an unborn child;
- If any clinical trial methods are used in the research.
- A promise to pay compensation in the event of injury or death resulting from a clinical trial for which subjects are entitled to compensation must be provided by the sponsor, whether it be a pharmaceutical company or an institution, along with the application for permission to conduct a clinical trial to the Licensing Authority as defined in clause (b) of Rule 21.
- The licencing authority may, after giving the sponsor a chance to demonstrate why such an order should not be passed, suspend or cancel the clinical trial if the sponsor fails to provide medical management for the subject’s injury, financial compensation for the trial subject for injury related to the clinical trial, or financial compensation to the subject’s nominee(s) in case of clinical trial related death of the subject.
Conclusion
There are optimistic rumours that forthcoming improvements to the regulatory environment would increase clinical trials in India. Simply described, a clinical trial is an investigation into the safety and efficacy of a novel therapy for a specific medical disease. In order to ensure patient safety throughout a clinical study, doctors must adhere to a strict timetable because little is known about the new treatment at that point. With the onset of globalisation, it is crucial to uphold international standards and best practises while respecting local customs and values and looking at creative methods to offer patients in India access to cheap healthcare. To foster innovation, regulators, business, and academics must work together. The regulatory framework must be reasonable and supported by data, not just conjecture.
Following several complaints of unethical procedures in the media, the CDSCO office tightened the rules for carrying out clinical studies in 2013. Numerous new rules were put into place, and many of them caused sponsors of global clinical trials (GCT) in India to become rather confused and hesitant. Changes were made to address issues with those regulations, but significant work remained, as shown by the fact that the number of clinical studies authorised by the Indian regulator has not yet reached levels seen before 2013.
The 2019 New Drugs & Clinical Trials Rules were enacted by CDSCO after reviewing the regulations for clinical trials and new drugs in order to address the issue of decreased clinical research in India.
“The ethical justification for undertaking health-related research involving humans is its scientific and social value: the prospect of generating the knowledge and the means necessary to protect and promote people’s health.” – Guideline 1, CIOMS, WHO, 2016.
Read Our Article: An Interplay Between CDSCO Guidelines & Public Health Safety









